
What is PET Drug Manufacturing?

The short half-life places obvious limitations on the manufacturing and dispensing of PET drugs. That said, the half-life of F-18 is sufficient to allow for the supply of F-18 PET drugs within a geographic area with a radius up to about five hours from the manufacturing facility. Although examples exist where distribution takes place over longer distances, this practice can result in significant costs due to transportation and product losses due to radioactive decay. It is also possible to manufacture and distribute PET drugs with shorter half-lives (e.g., C-11 with a half-life of 20 min), but this practice is limited to the smaller geographic areas.
Two subsets of manufacturers supply PET drugs in the US: (a) for on-site clinical use only“ (self-producers”) or (b) for broad clinical use (“commercial suppliers). The terminology varies, but self-producers are often thought of as “academic centers,” whereas wide geographic areas are served by commercial suppliers. Self-producers manufacture PET drugs for use within their own radiology or nuclear medicine departments (single location) and do not typically market to third-party PET imaging centers. On the other hand, commercial suppliers manufacture PET drugs from one or several locations for use by third-party PET imaging facilities.
Self-producers and commercial suppliers are often involved as contract manufacturers for innovators in the development and commercialization of new PET drugs. Since most innovators lack manufacturing capabilities for PET drugs, innovators must also rely on PET drug contract manufacturers for support during the FDA approval process and for commercial distribution after FDA approval.
Although practices vary, most common manufacturing processes yield the PET drug in a single multi-dose vial, which must be dispensed into a syringe for intravenous administration to a patient. The dispensing process takes place either in a nuclear pharmacy or a nuclear medicine department. The FDA is responsible for the regulatory oversight of the manufacturing process for the multi-dose vial, while state boards of pharmacy and/or medicine are responsible for the dispensing process into patient-specific doses. The prototypical workflow and regulatory landscape for PET drugs may be summarized as follows:
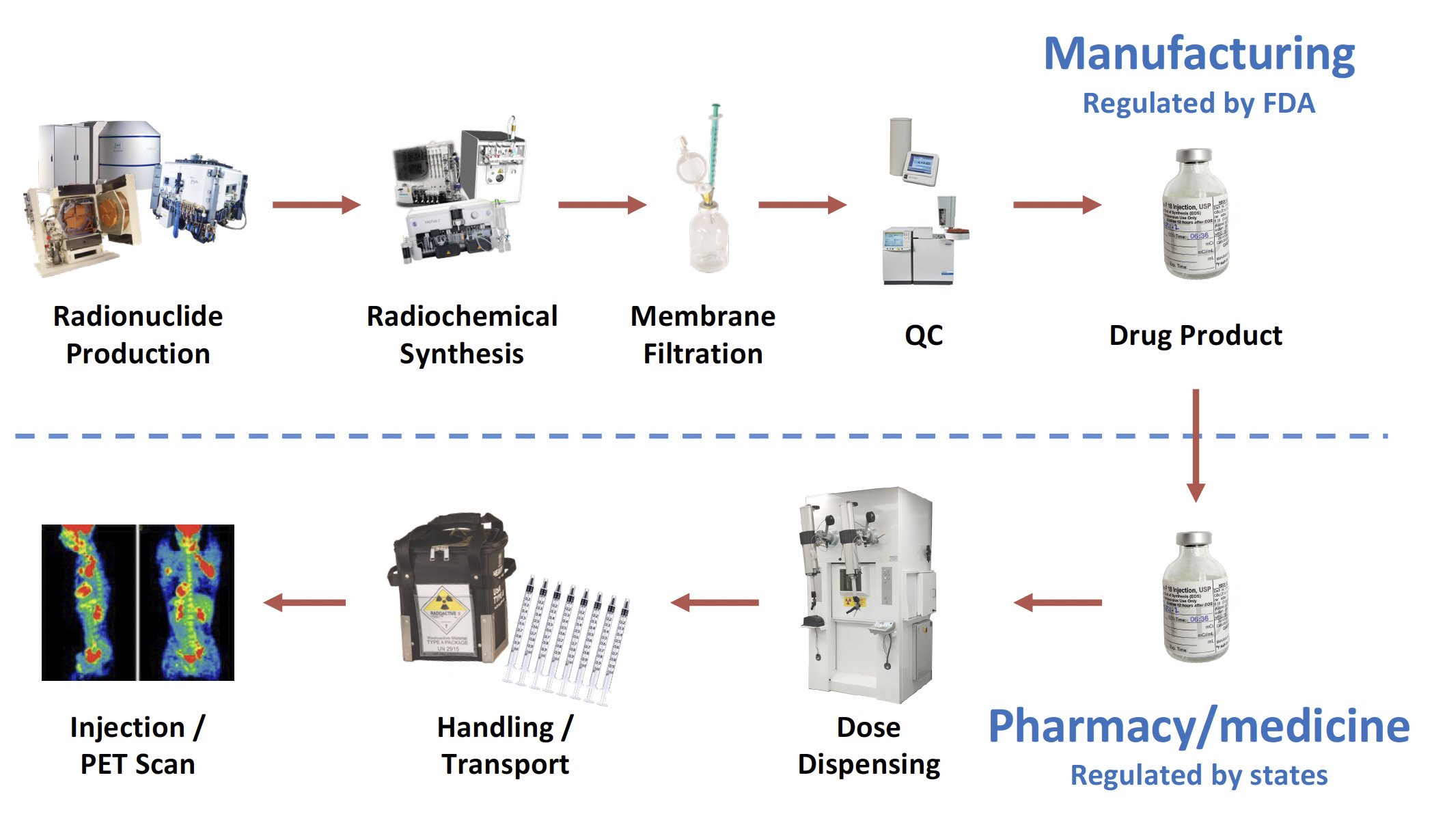
During all phases of handling and transportation, PET drugs are protected from accidental contamination. Simultaneously, operators are protected from the high energy radiation emitted by PET drugs. This requires the use of remote handling techniques and shielded hot cells, as well as shielded containers for transportation. This highlights a significant challenge for PET drugs: how to simultaneously protect operators from the high energy radiation emitted by PET drugs while protecting the drug itself from contamination that might compromise the safety of the PET drug.
What is PET Drug Dispensing?

The success of a PET drug means reaching patient within a very short shelf life, and a clear understanding of the respective roles played by drug manufacturers and nuclear pharmacies in North America, which is substantially different from other ICH territories such as the European Union.
In the US the FDA regulates manufacturing, which ends with the release of the drug product (typically a vial) to a health care professional (HCP). A HCP withdraws a patient-ready dose (PRD) from the vial into a syringe. It is critical to keep in mind that “the dispensing of patient doses […] is considered practice of pharmacy and not covered under the PET CGMP regulations” (FDA MAP 7356.002P).
The majority of US Healthcare Centers do not have on-site nuclear pharmacies and rely on 3rd party private nuclear pharmacies (stand-alone or network) for the preparation and dispensing of PRDs. The scientific justification of PRDs is developed and maintained by the sponsor and service provider (if involved), to support State Board of Pharmacy (BoP) inspections. The primary BoP information package includes syringe/container stability studies through shelf life, including Beyond Use Date (BUD) (within the manufactured vial and according to testing per the package insert) , as well as shipping studies for physical integrity (land/air shipment). Because these activities occur beyond manufacturing, they are typically not included into the drug product CTD (NDA/BLA).
The sponsor/manufacturer typically establishes a Master Services Agreement (MSA) and a Quality Assurance Agreement (QAA) with a pharmacy/network that governs the partie’s responsibilities regarding PRD preparation and distribution. A Clinical Trial Agreement (CTA) typically serves as MSA until drug regulatory approval.